Medical device PLM and quality: the cost of inconsistency


How document-first operating models create regulatory risk across the medical device lifecycle
- Inconsistency is a risk, both from a quality and business standpoint. Drift across labeling, IFUs, training, and submissions slows approvals and weakens audit readiness.
- The root cause is the operating model. High-risk statements get rebuilt across documents, so change does not propagate, and micro-variants multiply across teams, templates, and markets.
- The fix is to control content, not documents. Treat regulated statements as governed components with ownership, versioning, and traceability so that outputs and deliverables can scale without drift.
Problem and stakes
In regulated environments, consistency is of utmost importance. A single misaligned document or data point can spell trouble. Alignment is the baseline for regulatory review. Audits do not fail because they lack supporting documentation. They fail when labeling, packaging, and accompanying documents do not behave like a coherent, consistent, and defensible regulatory narrative.
Notified body assessments routinely become inefficient and take more time, at the cost of scalability. This happens when documentation is inconsistent across the technical file, labeling, and quality records. Source: EU guidance for notified body reviews.
The pattern is predictable: PLM defines the product and Quality defines the controls, but siloed teams still duplicate the same information across IFUs, labels, training, supplier instructions, and submission sections.
These rewrites trigger differences, that look insignificant and harmless at first sight, but each creates opportunity for the regulatory content to drift.
“A single missing update or inconsistent statement can lead to audit findings, fines, or worse.”
Source: Roadmap to better compliance.
A quick example:
- “Let’s eat, Grandma.”
- “Let’s eat Grandma.”
The comma determines the outcome for Grandma.
That example is playful. In medical devices, a comma can change meaning:
- “This is for internal use only.”
- “This is for internal use, only.”
In isolation, a reviewer may read them as the same. In a regulated documentation system, they are not.
That single comma creates a micro-variation. Micro-variations break exact matching, reduce translation reuse, and invite further edits as content moves between documents. Humans may gloss over them. AI-assisted checks will not. This is the cost of inconsistency: quality risk first, then time and money.
Scale makes drift inevitable. As variants, markets, and languages expand, small inconsistencies stop being edge cases and become a predictable failure mode under MDR, IVDR, and FDA quality system requirements.
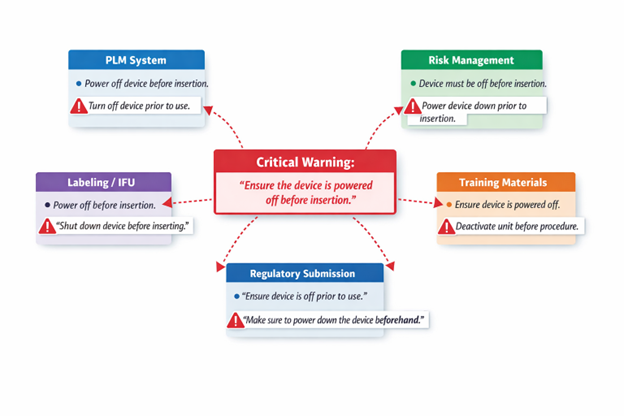
Impact on operations, risk, and growth
The consequences of inconsistency extend far beyond documentation. In large device manufacturers, product information is spread across Word files, spreadsheets, local databases, and enterprise systems, including PLM and quality platforms. When PLM, quality, and content repositories operate in silos, references drift.
When design data, risk assessments, and quality records don’t align, teams repeat work and hunt for discrepancies. Product development slows, not because engineering stalls, but because information cannot be trusted. Teams spend time answering questions they should not have to ask:
- Which version is authoritative?
- Which language copy is current?
- Which market variant was updated?
- Which document did not get the change?
In many traditional settings, teams copy and paste content across files, creating libraries of near-identical content with no single source of truth.
That friction creates visible business impact. Review cycles take longer and require more effort from SMEs to reconcile differences. This process becomes a bottleneck that makes change requests unscalable. At the process end, releases are delayed and late because every output had to be revalidated.
Notified bodies in the EU have been explicit about the consequences. Team-NB notes that assessments are delayed when information is hard to locate in poorly structured documentation, and it calls out inconsistency in duplicated device description information as a source of friction.
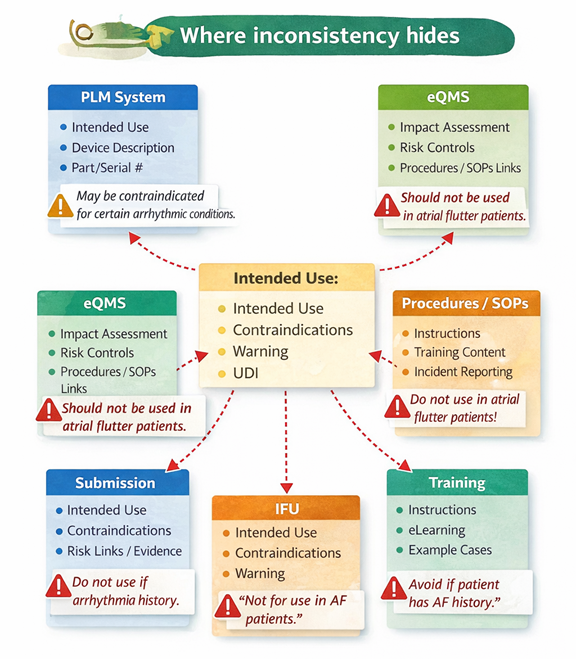
The same logic applies beyond the EU.
Regulators across regions expect controlled information, especially labeling and IFUs because they are linked to safety regulations. In the United States, document control is foundational under the Quality System Regulation (QSR). 21 CFR 820.40 requires procedures to control required documents. Source: eCFR, 21 CFR 820.40
This is about more than formality. When systems cannot reliably control or trace what is said, where it appears, and which version was released, quality risk multiplies. A requirement changes, but the verification reference stays old. A risk control changes, but the safety notification language does not. A CAPA identifies a documentation root cause, but training content does not reflect the corrective action in all languages. Each gap becomes a future audit finding, or a future field issue.
Real-world examples show how small labeling inconsistencies can become formal corrective actions. An FDA recall notice for a pressure-injectable catheter kit describes a mislabeling issue where the lid label indicated “non-coated,” while other packaging information indicated chlorhexidine-coated. Source: U.S. FDA Recall Pressure Injectable Catheter Kits for Mislabeling - EIF-000543 August 10, 2023
A risk for market competitivity and innovation
Inconsistencies slow approvals and market entry. Every delay shrinks the window of revenue and momentum. Late entry means lost sales and playing catch-up on pricing and share.
Medical devices scale through variants, configurations, and global market access, which means more outputs, more languages, and more compliance endpoints. MedTech Europe’s Facts & Figures 2025 report describes a sector of more than 38,000 medical technology companies in Europe, with about 90% SMEs, and a market size around €170 billion in 2024, projected globally to be ~$670 billion by 2027.
The ability to ensure the creation of consistent information is becoming a competitive differentiator. Those who cannot keep up face higher risk, slower approvals, and slower growth in an unforgiving market.

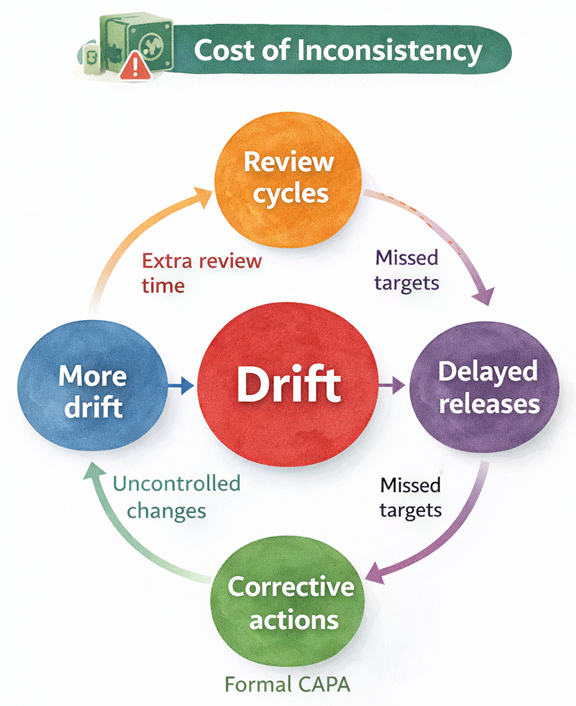
Why inconsistency persists
Inconsistent content is the predictable outcome of an operating model that treats documents as the unit of control.
Documents are easy to produce. They are hard to govern when the same information must appear across audiences, formats, markets, and long product lifecycles.
The problem is not documentation. It is what organizations mean when they say “document”. A document is treated as a linear artifact, approved as a whole, then reissued in full when changes occur. That model clashes with how regulated product information actually evolves across PLM, quality, and regulatory processes. The result is predictable: teams keep re-expressing the same controlled statements inside static files, and drift accumulates.
Three patterns show up almost everywhere:
- First, systems of record and systems of communication are disconnected. Structured truth lives in PLM, risk, quality, and tracking systems, while published content is assembled late and maintained manually.
- Second, ownership is fragmented. Multiple functions edit the same statements for different documents, and micro-variants multiply. Reviewers start accepting “close enough” because absolute alignment becomes too expensive to maintain.
- Third, change does not propagate reliably. Downstream teams cannot reliably identify every duplicated instance, especially across markets and translations.
Copy and paste is deceptively reassuring. It feels safe because it is familiar. But it turns controlled statements into manual transcription tasks. Even diligent teams can introduce errors through small, well-intended edits. These are invisible defects that survive review because they don’t look like defects.
In a content-first model, high-risk content is created once, governed once, and reused everywhere. Ownership, approval, versioning, and traceability are defined at the component level. Teams do not copy text. They reference it. The same approved statement can be assembled into IFUs, labeling, SOPs, training materials, and submission outputs, without diverging. Many outputs, one source.
When a change event occurs, the component is updated once and approved once. That change then propagates automatically to every document that uses it. Manual coordination drops dramatically. The process no longer depends on heroics or individual vigilance.
AI is not a shortcut
AI can accelerate content operations when the foundation is controlled and current. It does not fix inconsistency. It exposes it and compounds it at scale.
When content is fragmented and duplicated, AI moves fast on whatever it is given. It amplifies the drift.
In regulated environments, AI working across duplicated documents cannot reliably identify what is authoritative or trace an output back to approved design intent, risk controls, and evidence. That is why early AI initiatives in document-first environments often relocate effort rather than remove it. Review shifts downstream to release decisions, submissions, and inspections, where correction is most expensive.
When authoritative content exists as governed components with ownership, versioning, and traceability across PLM, quality, and regulatory records, AI can accelerate what is already reliable. It can support impact analysis, flag propagation, and scale multilingual delivery without weakening inspection defensibility.
The direction is clear: tighter alignment to standards, stronger process control, and greater scrutiny of how information is governed across the lifecycle. Without that change, automation increases exposure.
If you want speed without exposure, you need a content operating model built for control and reuse.
The solution: a structured, AI-ready approach
Many quality and compliance risks come down to how regulated statements are created, reused, and changed across the lifecycle. Document-centric approaches do not scale under MDR, FDA requirements, and AI-assisted workflows.
Instead of controlling documents, control the content used to assemble them.
A structured content approach treats regulated information as modular components, governed by ownership, approvals, traceability, and clear variant rules. It creates a single source of truth for statements that must never diverge and allows the organization to generate controlled outputs for each market, language, and audience.
Start by identifying the content that creates the highest risk when inconsistent: intended use, indications, contraindications, warnings, risk controls, key claims, device descriptions, UDI-related information, and training or procedures that implement controls.
Then design an information model for that content. Define the component types and the metadata that matters in regulated environments: product variant, market, language, audience, lifecycle stage, risk class, and status. This is where governance starts. If the model is weak, reuse will be weak, and inconsistency will return.
Once components exist, reuse becomes the default. In practice, this changes how high-risk statements are managed. It is a controlled component used wherever needed, in the exact approved form. When it changes, the change propagates across every output that uses it. That is how you remove cursor placement risk from the system.
Governance then becomes enforceable rather than aspirational. Every component has an owner, approvals, version history, release controls, and an audit trail built into the lifecycle.
Traceability turns this into a quality system advantage. When requirements, risks, and evidence are linked to the exact content that implements controls and communicates safe use, defensibility improves. Review accelerates because auditors can navigate from a claim or control to evidence, and to the user-facing expression of that control.
Multilingual scaling becomes manageable. Translate components, not entire document sets. Reuse increases and review focuses on changes, not full revalidation. All language versions stay anchored to the same controlled source.
This shift requires discipline, governance, and cross-functional alignment.
Expected outcomes
The value of a structured, governed content approach is practical and measurable. It shows up in fewer delays, fewer surprises, and faster, safer change management.
Operationally, the biggest wins come from change propagation. When a design change or a CAPA triggers an update, the system identifies where controlled content is used and drives updates through reuse, not manual editing. This reduces rework and shortens review loops.
On the risk side, the most valuable outcome is the elimination of invisible inconsistencies. When labeling and IFU content is managed as governed components, the likelihood of mismatched statements across packaging elements drops, and discrepancies become easier to detect before release.
On the growth side, the benefit is scalability. Global market access depends on being able to produce consistent, compliant outputs across markets and languages, repeatedly, over long product lifecycles. That scalability becomes a growth advantage when product families expand; regulations evolve, and change events keep coming.
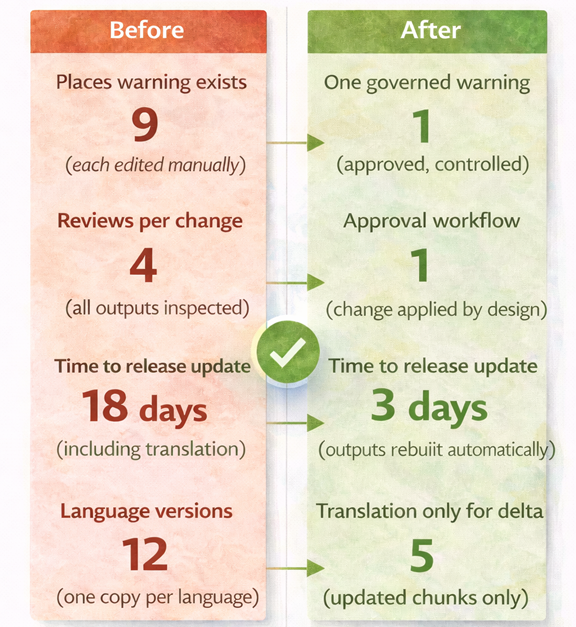
If you want to reduce drift, start small and make it measurable. Pick one device family and one set of high-risk statements. Map where they appear across PLM, risk, quality, labeling, IFUs, training, and submissions. Then refactor those statements into governed components with clear ownership and change control.
The goal is simple: many outputs, one truth.