
The biggest challenges that pharmaceutical organizations face when going to market are documentation complexity and regulatory variability. Dossiers count thousands of pages and demand a high level of complexity, but the regulatory landscape is also constantly shifting. From having different regulatory requirements based on regions and countries, AI- and technology-driven regulatory changes to legislative changes targeting the adjustment of macroeconomic forces, pharmaceutical companies are in for a ride.
According to the FDA’s official data, applications vary in size, reaching more than 150GB per submission. A New Drug Application (NDA) dossier can have from 1GB to more than 100GB, while a Biologics License Application (BLA) can have a size of over 168.62GB when compressed. 100GB can translate into hundreds of thousands of pages, depending of course, on file types. Either way, it is easy to understand this kind of volume can pose serious problems when managed without the right tools.
How many pages of text can fit in a gigabyte? How much is one gigabyte of data?
- Text files: Nearly 678,000 pages per gigabyte
- Emails: More than 100,000 pages
- Microsoft Word files: Almost 65,000 pages
- PowerPoint Slide Decks: Roughly 17,500 pages
- Images: Close to 15,500 pages
Market entry delay: inefficient regulatory content management
In a survey conducted by the European Medical Writers Association (EMWA), medical writers were asked what are main issues that are affecting the quality of the regulatory submission dossier. Their answers highlight the critical bottlenecks:
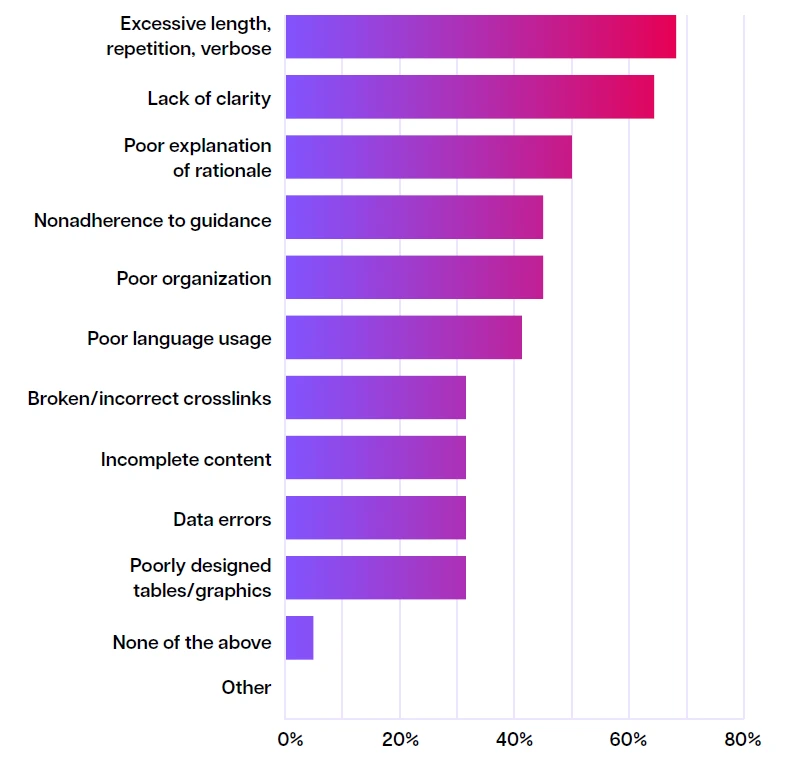
Top challenges in delivering high-quality regulatory files
1. Inconsistent use of terms
From clinical, non-clinical, CMS and safety, pharma teams are grappling with an abundance of regulatory documents authored by diverse groups with varying linguistic, professional and academic backgrounds. When they collaborate on a unitary dossier like the regulatory submission dossier, the challenge of inconsistent terminology arises, as each contributes using their own styles, reference materials and terminologies. As a result, considerable time is spent in internal and external meetings to compare documents and determine what certain terms mean and how to best align them, causing costly delays.
According to Norman R. Schmuff, from the Center for Drug Evaluation and Research (CDER), consistent terminology reduces regulatory submissions issues and delays, as it:
- “Eliminates confusion about synonyms, potentially synonymous terms
- Enables an ontology (i.e. properties and the relations between them
- Permits data analytics (e.g. how many assay procedures use CZE, for what classes of drugs)"
2. Traceability
Content within a dossier has interdependencies; for example, a Lab Manual or a Clinical Study Report (CSR) will contain overlapping information, taken from a Protocol document. This means that when content is updated in one of the documents, it must also be updated in all others. To keep track of these links between documents, teams need to have them mapped out. In a file-based approach, this mapping relies on manually created tables with entries that contain identification numbers representing various pieces of content. Each time a new interdependency is established, a new entry must be created and a new relationship between documents maintained. Because it’s easy to make a mistake or forget to make an update, this process isn’t sustainable and introduces significant risk.
3. Regulatory fragmentation
Different countries and regions maintain diverse regulatory frameworks for managing health data. For example, European countries enforce the GDPR, which imposes stringent requirements on personal data processing, while the US relies on HIPAA for patient privacy protection. Meanwhile, emerging regulations like South Africa’s POPIA and California’s CCPA add variability within global regions. This lack of harmonization creates complexities in collecting, sharing, and submitting clinical and real-world data as part of dossiers, often requiring region-specific adaptations.
These geographical regulatory variations force pharma companies to adapt their regulatory submission strategies, often preparing separate dossier versions tailored to local data requirements, formats, and validation standards. Managing multiple submission versions increases complexity, time, and cost while introducing risks of inconsistencies. Additionally, innovators in digital health and personalized medicine face growing regulatory uncertainty, as evolving local rules lag behind technology advances.
4. Inefficient update and quality control of TLF (in-text tables, listings and figures)
TLFs are essential, derived from various data sources and are used to summarize and analyze datasets of a clinical study into an understandable format. They are used to answer regulatory questions and present clinical evidence.
Although the process may seem straightforward, it demands frequent, complex quality checks to verify the validity of the information included. When medical writers ask for changes to TLFs, this triggers a series of steps:
- Medical writer requests a table modification
- Statistical programmer rewrites code
- Quality control re-validates (full process)
- Biostatistician re-reviews
- Process repeats if further changes are needed
The non-automated, manual TLF authoring leads to incomplete content, data errors or non-adherence to guidance issues such as:
- Out-of-Specification (OOS) and Out-of-Trend (OOT) rates for TLF data
- Invalidated OOS Rate (IOOSR): a KPI that indicates poor initial TLF validation
- Data entry lag time: time between patient visit and data entry - TLF misalignment with source data often stems from data timing mismatches
Automated content reuse – the answer to your regulatory delays
How many thousands of documents in your organization might need to use the same chunks of content? What happens when an author can't find the specific content they want to reuse, or doesn't know it already exists? Typically, they write or rewrite it.
If they do manage to find the chunk they want, they might copy the source and paste it into the new document. But if there's no connection between the two versions – the source and the copy-pasted instance, they can soon get out of sync, potentially leading to accuracy or compliance issues. If changes are made to the original chunk, how do you ensure they're reflected in the copied chunk?
Some authoring teams manually track which content chunks have been reused where and, when the original content gets updated, they make the same updates to all the copies. But if you're working with large sets of documentation or multiregional and multilingual content, things can quickly get out of control.
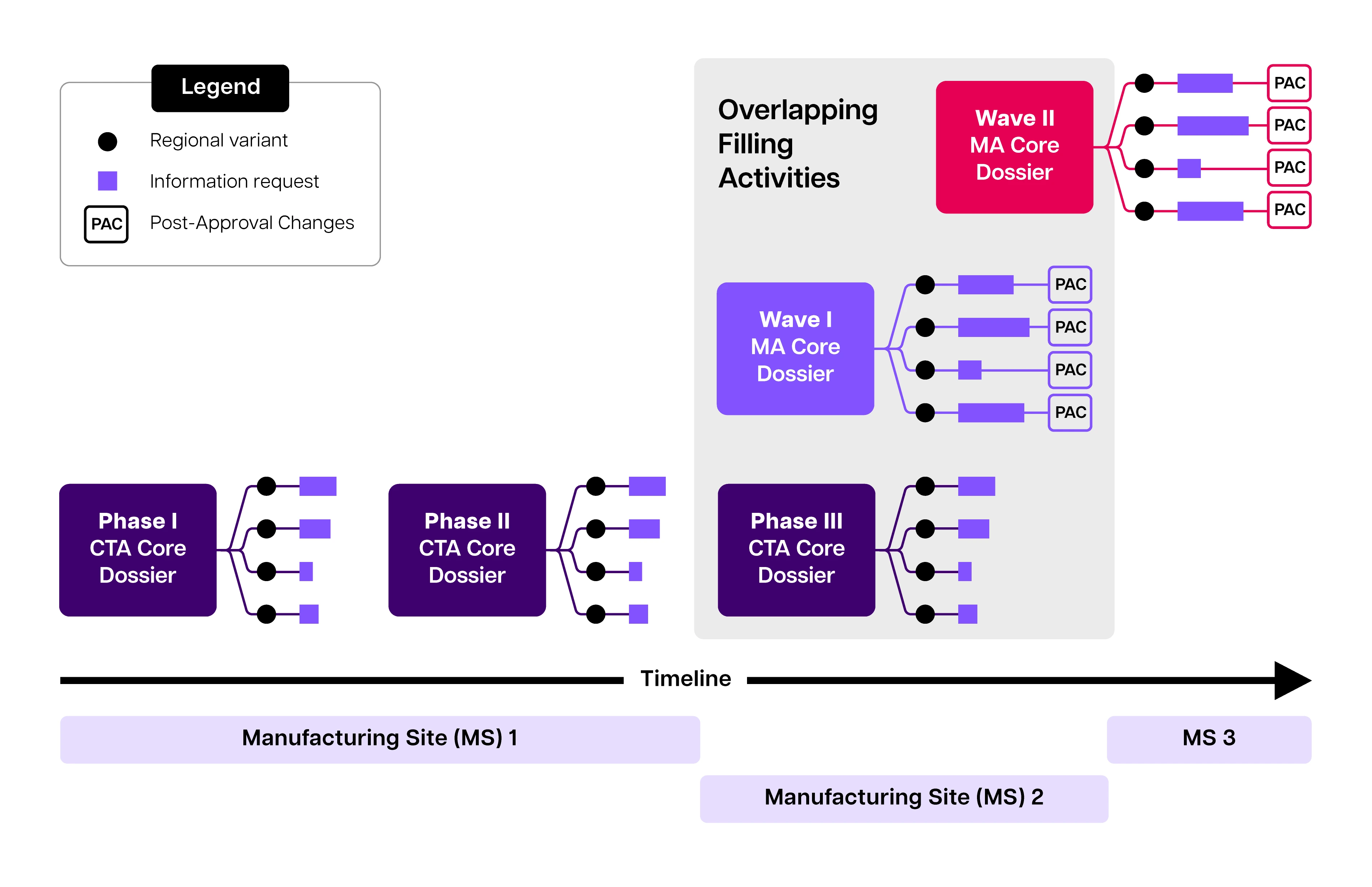
Compounding submission volume and complexity during development. The quantity of dossiers, country variants, information requests, and manufacturing changes substantially increases over time. For accelerated programs, later stages of development are often characterized by overlapping milestones and filing expectations, wherein the phase 3 trial may overlap with the filing of the marketing application. The marketing application may be filed in multiple discrete “waves” including groupings of different regions, wherein authoring, review, and approval timelines may overlap
Source: Structured content and data management-enhancing acceleration in drug development through efficiency in data exchange - PubMed
This way of working introduces inconsistencies, making it difficult for reviewers, internal or external, to analyze the submissions dossier, in more serious cases leading to the rejection of the submission.
If we think of copying and pasting content chunks as uncontrolled content reuse, then the alternative has to be controlled content reuse. So, what does that look like?
What is controlled content reuse?
Controlled content reuse allows a content chunk (or component) to be created once and used anywhere it's needed without being copied. It sounds simple, but the impact can be huge. As a key aspect of structured content authoring (SCA), controlled content reuse enables the reuse of any vetted content component anywhere, without having to copy/paste or otherwise duplicate it. In an SCA environment, authors produce content in small chunks that can be easily managed, kept up to date and reused in a controlled way.
A component content management system (CCMS), also known as a structured content management system (SCMS), or a Structured Content and Data Management System (SCDM), is the technology that lets you create and manage structured content components and reuse them in a controlled way.
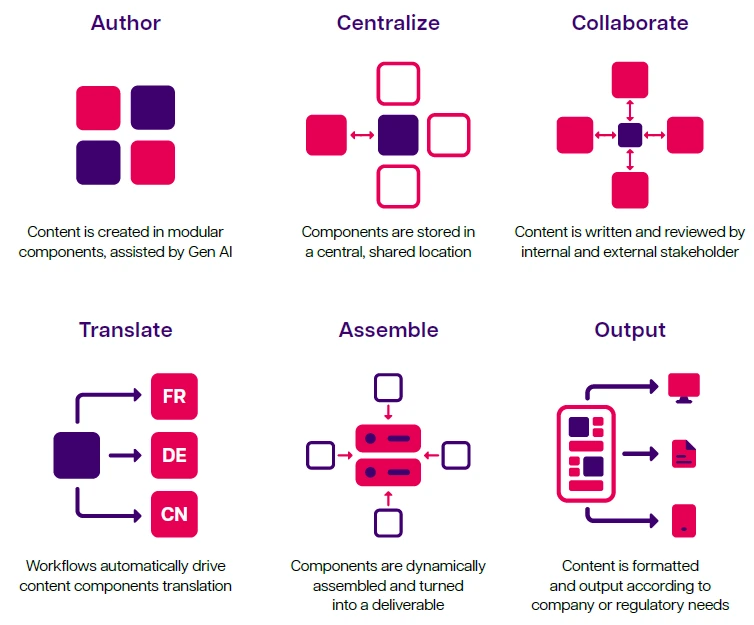
The controlled content reuse approach
Verbatim vs derivative content reuse
Verbatim content reuseAt this stage, you may think that content reuse revolves around reusing copy chunks with no ability to modify them.
This is certainly one type of content reuse. In the context of structured content authoring with a CCMS, reusing unchanged content chunks is known as verbatim content reuse (or locked reuse). This is useful when information needs to be repeated ‘as is’ within or across documents and other assets, such as those that contain legal information, health and safety provisions, disclaimers or chemical substance compositions, to name just a few.
Best practice is to keep these locked chunks as small and specific as possible to simplify their integration into various documents and assets.
Verbatim content reuse allows:
- Content to be updated quickly across various assets
- Easy tracking and control of reused content
- Documents to follow agreed standards and formats
- Enhanced consistency, accuracy and uniformity of content
To help ensure a high level of content reuse, most organizations allow derivative content reuse, where content chunks aren't locked and can therefore be edited by authors. Making edits creates a new chunk of content. Authors reuse derivative content to create new documents from content components that already exist, modified to suit the new document or other asset.
The newly created (or derivative) content chunk is a ‘child’ of the ‘parent content’ or original chunk. This parent-child relationship is recorded in a part of the system, called Component Content Management System (CCMS), which notifies the author of the derivative content if the original content gets modified. The author can then decide whether or not to incorporate the changes into the derivative content.
Derivative content can be created using content variables and conditions in your publication, or by editing content manually. A translation is also considered to be derivative content.
Derivative content reuse allows:
- Quick authoring of content across documents with different purposes and audiences
- More flexibility for authors to adapt the content without loss of control
- Long-term consistency by tracking the relationship between the original and the derivative content
The future of regulatory submissions content: data-driven submissions
In pharmaceutical regulatory submission documentation, there’s a wide variety of stakeholders, ranging from medical writers to regulatory bodies to medical staff and patients. They all have different expectations and needs, making the process too complex to be managed sustainably in the traditional document-centric way. We have seen in the previous paragraphs of this article that content from one document is interrelated with multiple other documents, making changes, adaptations, reviews and approval cumbersome.
To solve this, a broader, more fundamental shift in the documentation process is required.
Sponsors need to revisit the entire notion of working with documents. The success and speed of documentation processes depend on contributors’ ability to access and interpret reliable data and extract relevant points for the documents they create or review. So, the first logical step in a pharma organization’s evolution is to start treating documents as data.
This direction is endorsed by a considerable number of initiatives and regulatory standards. Industry stakeholders understand the need for joining forces to make documentation authoring and review easier, so that innovative drugs reach the market faster.
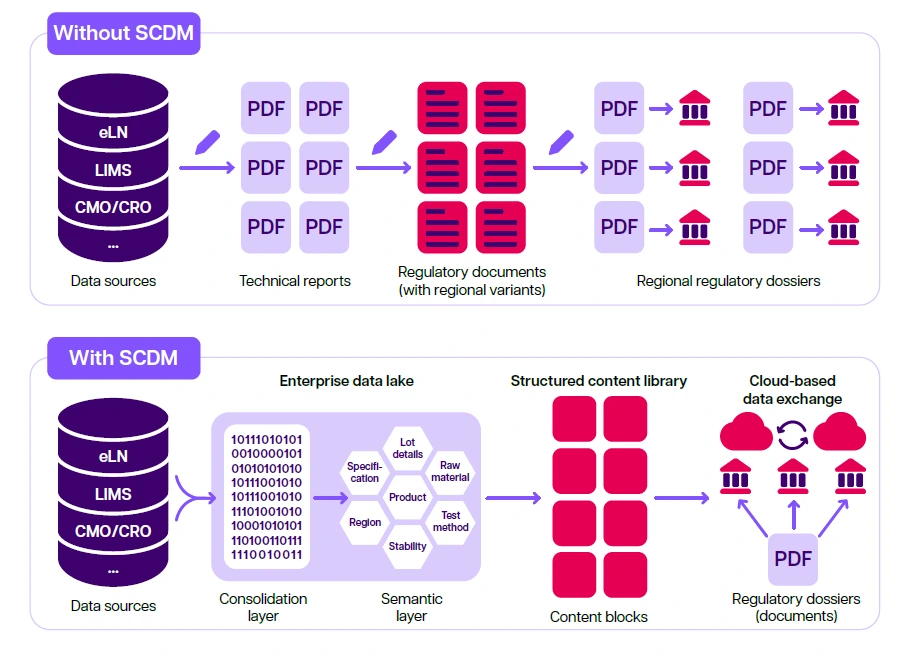
Comparison of regulatory submission processes with and without SCDM. A comparison of regulatory CMC submission assembly and dispatch is shown. Without SCDM, data is manually transcribed from the source into a technical report. The technical report is then used as a source document for a regulatory document, of which there are multiple regional variants created manually. Compilations of documents are then sent individually to health authorities for review, requiring the company to maintain multiple regional dossier variants. With SCDM, manual authoring steps can be reduced or eliminated as content and data can be taken directly from the source system, processed and semantically mapped via an enterprise data lake, and made available for incorporation in a regulatory filing via the structured content and data library, which is made up of content blocks. The content blocks can be reorganized, updated, and customized according to regulatory objectives and regional filing needs. The structured content and data library can achieve flexible output options, including electronic data submission, cloud-based information transfer, and printable paper-based filings.

Author
Todd Georgieff
Industry Lead for Pharma